8. Detection of binding events from ChIP-exo data¶
The workflow is comprised of five steps, as shown in next table. The first step, sample download and quality control, is for downloading samples from the NCBI SRA database, if necessary, or for executing the pre-processing quality control tools on all samples. After that, sample trimming and alignment are executed. Post-processing quality control on the ChIP-exo samples is performed, using Phantompeakqualtools. After that, the peak calling is performed, using MACE. Finally, DNA motif annotation is performed with the MEME Suite.
| Step | Jupyter Notebook | Workflow CWL | Tool | Input | Output | CWL Tool |
|---|---|---|---|---|---|---|
| Sample Download and Quality Control | 01 - Pre-processing QC | download_quality_control.cwl | SRA-Tools | SRA accession | Fastq | fastq-dump.cwl |
| FastQC | Fastq | FastQC HTML and Zip | fastqc.cwl | |||
| Trimming | 02 - Samples trimming | Trimmomatic | Fastq | Fastq | ||
| Alignments and Quantification | 03 - Alignments | chip-seq-alignment.cwl | BWA | Fastq | BAM | bwa-mem.cwl |
| Samtools | SAM | BAM Sorted BAM BAM index BAM stats BAM flagstats |
||||
Peak Calling and IDR |
04 - Peak Calling | peak-caller-MACE-SE.cwl | IGVtools | Sorted BAM | TDF | igvtools-count.cw |
| MACE | tagAlign | Peaks (TSV), plots | mace.cwl | |||
| BAMScale | BAM | TPM values | ||||
| DNA motif identification | 05 - MEME Motif | MEME Suite | Peaks | DNA Motif , plots | meme-chip.cwl |
8.1. Input requirements¶
The input requirement for the ChIPexo pipeline is the Sample sheet file.
8.2. Pipeline command line¶
The ChIPexo based project can be created using the following command line:
localhost:~> pm4ngs-chipexo
usage: Generate a PM4NGS project for ChIPexo data analysis [-h] [-v]
--sample-sheet
SAMPLE_SHEET
[--config-file CONFIG_FILE]
[--copy-rawdata]
Options:
- sample-sheet: Sample sheet with the samples metadata
- config-file: YAML file with configuration for project creation
- copy-rawdata: Copy the raw data defined in the sample sheet to the project structure. (The data can be hosted locally or in an http server)
8.3. Creating the Detection of binding events from ChIP-exo data project¶
The pm4ngs-chipexo command line executed with the --sample-sheet option will let you type the different variables required for creating and configuring the project. The default value for each variable is shown in the brackets. After all questions are answered, the CWL workflow files will be cloned from the github repo ncbi/cwl-ngs-workflows-cbb to the folder bin/cwl.
localhost:~> pm4ngs-chipexo --sample-sheet my-sample-sheet.tsv
Generating ChIP-exo data analysis project
author_name [Roberto Vera Alvarez]:
email [veraalva@ncbi.nlm.nih.gov]:
project_name [my_ngs_project]:
dataset_name [my_dataset_name]:
is_data_in_SRA [None]:
Select sequencing_technology:
1 - single-end
2 - paired-end
Choose from 1, 2 [1]:
create_demo [y]:
number_spots [5000000]:
organism [human]:
genome_name [hg38]:
genome_dir [hg38]:
aligner_index_dir [hg38/BWA]:
genome_fasta [hg38/genome.fa]:
genome_gtf [hg38/genes.gtf]:
genome_chromsizes [hg38/genome.sizes]:
use_docker [y]:
max_number_threads [16]:
Cloning Git repo: https://github.com/ncbi/cwl-ngs-workflows-cbb to /Users/veraalva/my_ngs_project/bin/cwl
Updating CWLs dockerPull and SoftwareRequirement from: /Users/veraalva/my_ngs_project/requirements/conda-env-dependencies.yaml
bamscale with version 0.0.3 update image to: quay.io/biocontainers/bamscale:0.0.3--ha85820d_0
/Users/veraalva/my_ngs_project/bin/cwl/tools/bamscale/bamscale-docker.yml with old image replaced: quay.io/biocontainers/bamscale:0.0.5--h18f8b1d_1
bedtools with version 2.29.2 update image to: quay.io/biocontainers/bedtools:2.29.2--hc088bd4_0
/Users/veraalva/my_ngs_project/bin/cwl/tools/bedtools/bedtools-docker.yml with old image replaced: quay.io/biocontainers/bedtools:2.28.0--hdf88d34_0
bioconductor-diffbind with version 2.16.0 update image to: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_0
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/deseq2-pca.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/macs-cutoff.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/dga_heatmaps.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/diffbind.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/edgeR-2conditions.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/volcano_plot.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/readQC.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/R/deseq2-2conditions.cwl with old image replaced: quay.io/biocontainers/bioconductor-diffbind:2.16.0--r40h5f743cb_2
bwa with version 0.7.17 update image to: quay.io/biocontainers/bwa:0.7.17--hed695b0_7
/Users/veraalva/my_ngs_project/bin/cwl/tools/bwa/bwa-docker.yml with old image replaced: quay.io/biocontainers/bwa:0.7.17--h84994c4_5
There is not biocontainer image for gffread version 0.12.1
homer with version 4.11 update image to: quay.io/biocontainers/homer:4.11--pl526h9a982cc_2
/Users/veraalva/my_ngs_project/bin/cwl/tools/homer/homer-docker.yml with old image replaced: quay.io/biocontainers/homer:4.11--pl526h2bce143_2
mace with version 1.2 update image to: quay.io/biocontainers/mace:1.2--py27h99da42f_0
/Users/veraalva/my_ngs_project/bin/cwl/tools/mace/mace-docker.yml with old image replaced: quay.io/biocontainers/mace:1.2--py27h99da42f_1
meme with version 5.1.1 update image to: quay.io/biocontainers/meme:5.1.1--py37pl526h072abfd_3
/Users/veraalva/my_ngs_project/bin/cwl/tools/meme/meme-docker.yml with old image replaced: quay.io/biocontainers/meme:5.1.1--py27pl526h53063a7_3
Copying file /Users/veraalva/Work/Developer/Python/pm4ngs/pm4ngs-chipexo/example/pm4ngs_chipexo_demo_sample_data.csv to /Users/veraalva/my_ngs_project/data/my_dataset_name/sample_table.csv
6 files loaded
Using table:
sample_name file condition replicate
0 SRR4011416 Exp_O2_growth_no_rifampicin 1
1 SRR4011417 Exp_O2_growth_no_rifampicin 2
2 SRR4011421 Exp_O2_growth_rifampicin 1
3 SRR4011425 Exp_O2_growth_rifampicin 2
4 SRR4011418 Stat_02_growth_no_rifampicin 1
5 SRR4011419 Stat_02_growth_no_rifampicin 2
Done
The pm4ngs-chipexo command line will create a project structure as:
.
├── LICENSE
├── README.md
├── bin
│ └── cwl
├── config
│ └── init.py
├── data
│ └── my_dataset_name
├── doc
├── index.html
├── notebooks
│ ├── 00 - Project Report.ipynb
│ ├── 01 - Pre-processing QC.ipynb
│ ├── 02 - Samples trimming.ipynb
│ ├── 03 - Alignments.ipynb
│ ├── 04 - Peak Calling.ipynb
│ └── 05 - MEME Motif.ipynb
├── requirements
│ └── conda-env-dependencies.yaml
├── results
│ └── my_dataset_name
├── src
└── tmp
12 directories, 11 files
Note
ChIPexo based project variables
- author_name:
Default: [Roberto Vera Alvarez]
- email:
Default: [veraalva@ncbi.nlm.nih.gov]
- project_name:
Name of the project with no space nor especial characters. This will be used as project folder's name.
Default: [my_ngs_project]
- dataset_name:
Dataset to process name with no space nor especial characters. This will be used as folder name to group the data. This folder will be created under the data/{{dataset_name}} and results/{{dataset_name}}.
Default: [my_dataset_name]
- is_data_in_SRA:
If the data is in the SRA set this to y. A CWL workflow to download the data from the SRA database to the folder data/{{dataset_name}} and execute FastQC on it will be included in the 01 - Pre-processing QC.ipynb notebook.
Set this option to n, if the fastq files names and location are included in the sample sheet.
Default: [y]
- Select sequencing_technology:
Select one of the available sequencing technologies in your data.
Values: 1 - single-end, 2 - paired-end
- create_demo:
If the data is downloaded from the SRA and this option is set to y, only the number of spots specified in the next variable will be downloaded. Useful to test the workflow.
Default: [y]: y
- number_spots:
Number of sport to download from the SRA database. It is ignored is the create_demo is set to n.
Default: [1000000]
- organism:
Organism to process, e.g. human. This is used to link the selected genes to the NCBI gene database.
Default: [human]
- genome_name:
Genome name , e.g. hg38 or mm10.
Default: [hg38]
- genome_dir:
Absolute path to the directory with the genome annotation (genome.fa, genes.gtf) to be used by the workflow or the name of the genome.
If the name of the genome is used, PM4NGS will include a cell in the 03 - Alignments.ipynb notebook to download the genome files. The genome data will be at data/{{dataset_name}}/{{genome_name}}/
Default: [hg38]
- aligner_index_dir:
Absolute path to the directory with the genome indexes for BWA.
If {{genome_name}}/BWA is used, PM4NGS will include a cell in the 03 - Alignments.ipynb notebook to create the genome indexes for BWA.
Default: [hg38/BWA]
- genome_fasta:
Absolute path to the genome fasta file
If {{genome_name}}/genome.fa is used, PM4NGS will use the downloaded fasta file.
Default: [hg38/genome.fa]
- genome_gtf:
Absolute path to the genome GTF file
If {{genome_name}}/genes.gtf is used, PM4NGS will use the downloaded GTF file.
Default: [hg38/gene.gtf]
- genome_chromsizes:
TSV file with the genome chromosomes name and size.
If {{genome_name}}/genome.sizes is used, PM4NGS will use the downloaded file.
Default: [hg38/genome.sizes]
- use_docker:
Set this to y if you will be using Docker. Otherwise Conda needs to be installed in the computer.
Default: [y]
- max_number_threads:
Number of threads available in the computer.
Default: [16]
8.4. Jupyter server¶
PM4NGS uses Jupyter as interface for users. After project creation the jupyter server should be started as shown below. The server will open a browser windows showing the project's structure just created.
localhost:~> jupyter notebook
8.5. Data processing¶
Start executing the notebooks from 01 to 05 waiting for each step completion. The 00 - Project Report.ipynb notebook can be executed after each notebooks to see the progress in the analysis.
8.6. CWL workflows¶
8.6.1. SRA download and QC workflow¶
This CWL workflow is designed to download FASTQ files from the NCBI SRA database using fastq-dump and then, execute fastqc generating a quality control report of the sample.
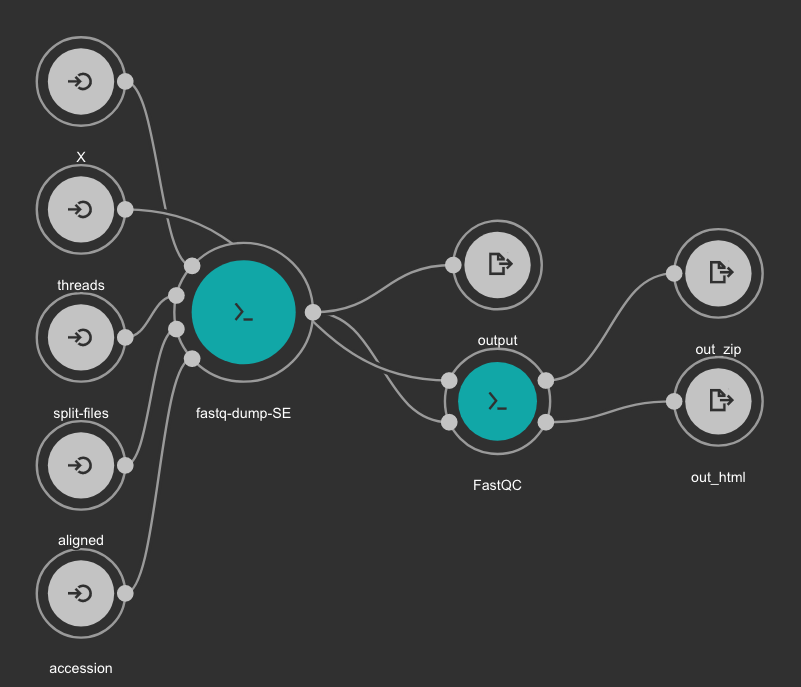
Inputs
- accession: SRA accession ID. Type: string. Required.
- aligned: Used it to download only aligned reads. Type: boolean. Optional.
- split-files: Dump each read into separate file. Files will receive suffix corresponding to read number. Type: boolean. Optional.
- threads: Number of threads. Type: int. Default: 1. Optional.
- X: Maximum spot id. Optional.
Outputs
- output: Fastq files downloaded. Type: File[]
- out_zip: FastQC report ZIP file. Type: File[]
- out_html: FastQC report HTML. Type: File[]
8.6.2. Samples Trimming¶
Our workflows uses Trimmomatic for read trimming. The Jupyter notebooks uses some basic Trimmomatic options that need to be modified depending on the FastQC quality control report generated for the sample.
8.6.3. BWA based alignment and quality control workflow¶
This workflow use BWA as base aligner. It also use SAMtools and bedtools for file conversion and statistics report. Finally, Phantompeakqualtools is used to generate quality control report for the processed samples.
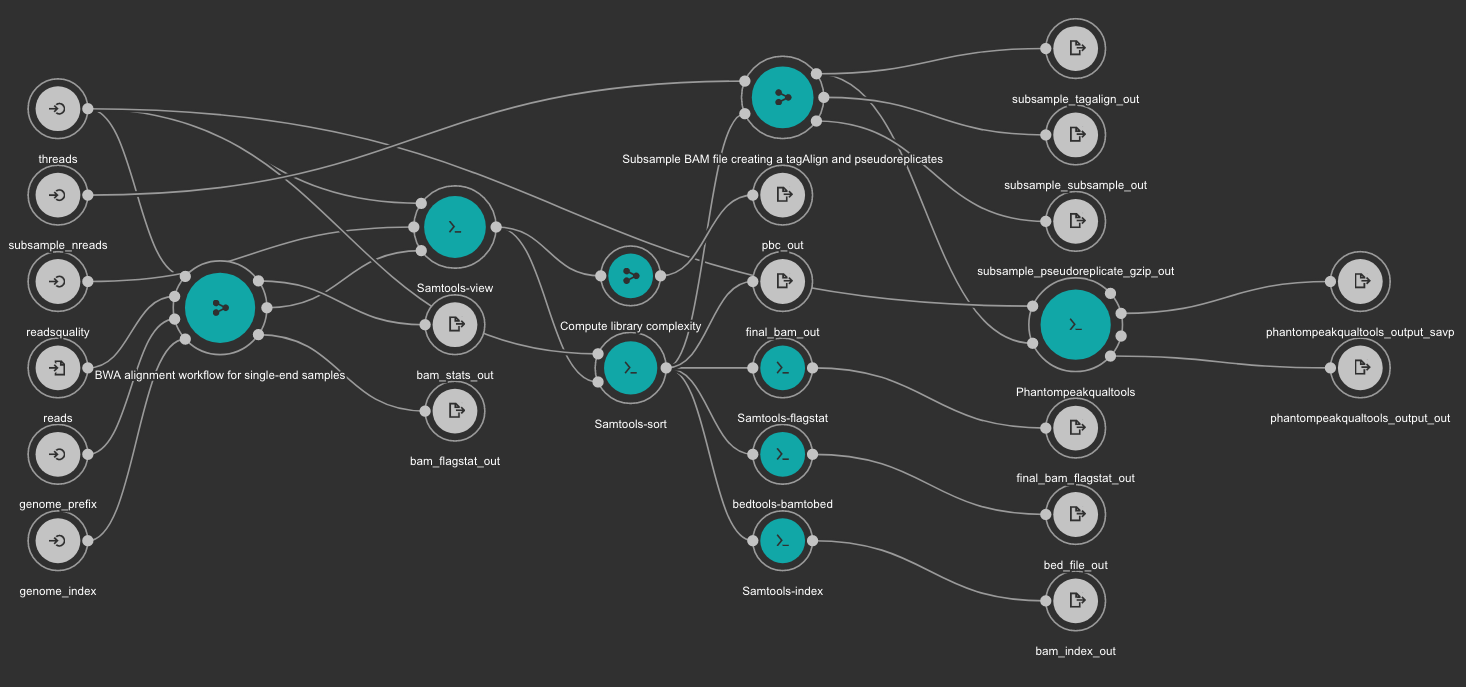
Inputs
- genome_index: Aligner indexes directory. Type: Directory. Required. Variable ALIGNER_INDEX in the Jupyter Notebooks.
- genome_prefix: Prefix of the aligner indexes. Generally, it is the name of the genome FASTA file. It can be used as os.path.basename(GENOME_FASTA) in the Jupyter Notebooks. Type: string. Required.
- readsquality: Minimum MAPQ value to use reads. We recommend for ChIP_exo dataa value of: 30. Type: int. Required.
- threads: Number of threads. Type: int. Default: 1. Optional.
- subsample_nreads: Number of reads to be used for the subsample. We recomend for ChIP_exo dataa value of: 500000. Type: int. Required.
- reads: FastQ file to be processed. Type: File. Required.
Outputs
- bam_flagstat_out: SAMtools flagstats for unfiltered BAM file. Type: File.
- bam_stats_out: SAMtools stats for unfiltered BAM file. Type: File.
- final_bam_flagstat_out: SAMtools flagstats for filtered BAM file. Type: File.
- bed_file_out:: Aligned reads in BED format. Type: File.
- final_bam_out: Final BAM file filtered and sorted. Type: File.
- bam_index_out: BAM index file. Type: File.
- pbc_out: Library complexity report. Type: File.
- phantompeakqualtools_output_out: Phantompeakqualtools main output. Type: File.
- phantompeakqualtools_output_savp: Phantompeakqualtools SAVP output. Type: File.
- subsample_pseudoreplicate_gzip_out: Subsample pseudoreplicates tagAlign gzipped. Type: File[].
- subsample_tagalign_out: Subsample tagAlign gzipped. Type: File[].
- subsample_subsample_out: Subsample shuffled tagAlign gzipped. Type: File[].
8.6.4. Peak caller workflow using MACE¶
This workflow uses MACE as peak caller tool. The annotation is created from the GTF file using a in-house python script available here. BAMscale is used for the quantification of the resulting peaks.
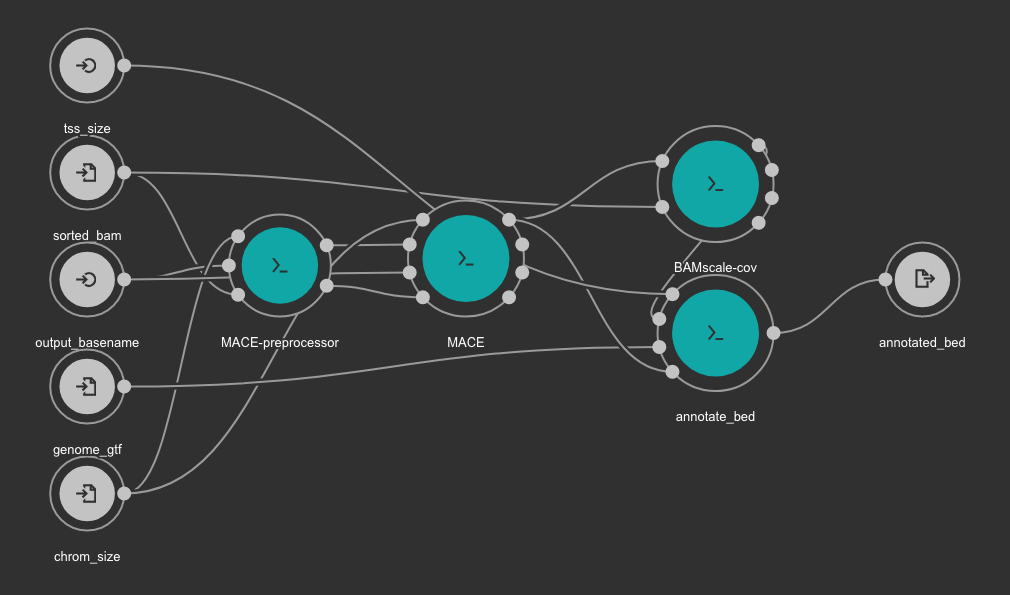
Inputs
- chrom_size: Chromosome size file. Tab or space separated text file with 2 columns: first column is chromosome name, second column is size of the chromosome. Type: File. Required. Variable GENOME_CHROMSIZES in the Jupyter Notebooks.
- output_basename: Prefix for the output file. Type: string. Required.
- genome_gtf: Genome GTF file. Variable GENOME_GTF in the Jupyter Notebooks. Type: File. Required.
- tss_size: Number of bp to use for TSS definition. We recommend use 1000. Type: int. Required.
Outputs
- annotated_bed: Annotated detected peaks in BED format. Type: File
8.6.5. MEME Motif detection workflow¶
Motif detection is executed using the MEME suite.
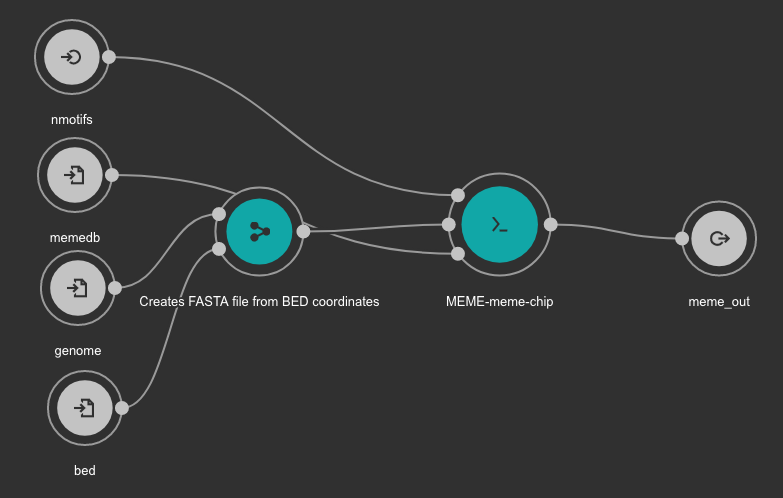
Inputs
- bed: BED file with detected peaks. Type: File. Required.
- memedb: MEME database for use by Tomtom and CentriMo. Type: File. Required.
- genome: Genome FASTA file. Variable GENOME_FASTA in the Jupyter Notebooks. Type: File. Required.
- nmotifs: Maximum number of motifs to find. We recommend use 10. Type: int. Required.
Outputs
- meme_out: MEME output directory. Type: Directory
8.6.6. MEME databases¶
MEME workflow depends on the MEME databases. Go to the MEME Suite Download page: http://meme-suite.org/doc/download.html
Download the latest version for the Motif Databases and GOMo Databases.
The downloaded files should be uncompressed in a directory data/meme. The final directory should be:
localhost:~> cd data
localhost:~> mkdir meme
localhost:~> cd meme
localhost:~> wget http://meme-suite.org/meme-software/Databases/motifs/motif_databases.12.18.tgz
localhost:~> wget http://meme-suite.org/meme-software/Databases/gomo/gomo_databases.3.2.tgz
localhost:~> tar xzf motif_databases.12.18.tgz
localhost:~> tar xzf gomo_databases.3.2.tgz
localhost:~> rm gomo_databases.3.2.tgz motif_databases.12.18.tgz
localhost:~> tree -d
.
├── gomo_databases
└── motif_databases
├── ARABD
├── CIS-BP
├── CISBP-RNA
├── ECOLI
├── EUKARYOTE
├── FLY
├── HUMAN
├── JASPAR
├── MALARIA
├── MIRBASE
├── MOUSE
├── PROKARYOTE
├── PROTEIN
├── RNA
├── TFBSshape
├── WORM
└── YEAST
19 directories
See also an example in our test project: https://ftp.ncbi.nlm.nih.gov/pub/pm4ngs/examples/chipexo-single/data/
8.7. Demo¶
PM4NGS includes a demo project that users can use to test the framework. It is pre-configured to use Docker as execution environment.
The ChIPexo based demo process samples from the BioProject PRJNA338159.
Use this command to create the project structure in your local computer
localhost:~> pm4ngs-chipexo-demo
Once it finish, start the jupyter server and execute the notebooks as it is indicated on them
localhost:~> jupyter notebook
[I 14:12:52.956 NotebookApp] Serving notebooks from local directory: /home/veraalva
[I 14:12:52.956 NotebookApp] Jupyter Notebook 6.1.4 is running at:
[I 14:12:52.956 NotebookApp] http://localhost:8888/?token=eae6a8d42ad12d6ace23f5d0923bcec14d0f798127750122
[I 14:12:52.956 NotebookApp] or http://127.0.0.1:8888/?token=eae6a8d42ad12d6ace23f5d0923bcec14d0f798127750122
[I 14:12:52.956 NotebookApp] Use Control-C to stop this server and shut down all kernels (twice to skip confirmatio
n).
[C 14:12:52.959 NotebookApp]
To access the notebook, open this file in a browser:
file:///home/veraalva/.local/share/jupyter/runtime/nbserver-23251-open.html
Or copy and paste one of these URLs:
http://localhost:8888/?token=eae6a8d42ad12d6ace23f5d0923bcec14d0f798127750122
or http://127.0.0.1:8888/?token=eae6a8d42ad12d6ace23f5d0923bcec14d0f798127750122
The results of this analysis is https://ftp.ncbi.nlm.nih.gov/pub/pm4ngs/examples/chipexo-single/